Ca fait longtemps que je voulais faire ce sujet en vidéo, je l’avais d’ailleurs traité dans mon livre « Insoluble mais vrai ! » consacré aux grands problèmes ouverts de la science. L’actualité m’a devancé avec la publications des résultats de la compétition CASP, et l’incroyable performance de l’algorithme AlphaFold de DeepMind.
Ce genre de sujet est assez compliqué à traiter en profondeur, car on y touche à plusieurs champs disciplinaires : la biologie évidemment, la chimie, la physique et maintenant l’IA. Pour ceux qui seraient encore curieux, voici quelques compléments.
Les protéines c’est compliqué
Parlons pour commencer des protéines. Pour illustrer mon propos j’ai pas mal hésité entre plusieurs protéines. Je voulais quelque chose de pas trop gros mais qui soit quand même parlant. J’ai initialement pensé à l’insuline, mais elle a deux inconvénients : elle est en fait formée de deux chaines (reliées par des ponts disulfure), donc mon propos était un peu moins clair. Et en plus elle n’a « que » 51 acides aminés. Or pour distinguer les « petits peptides » des protéines, il y a une limite arbitrairement fixée à 100 A.A., donc officiellement l’insuline n’est pas une protéine. En fait on s’en fout, mais je sais qu’il y a toujours quelques grincheux au fond qui attendent avec impatience que je dise un truc du genre « l’insuline est une protéine » 🙂
Mais rassurez-vous, j’ai dit plein d’autres trucs faux. Par exemple ce que j’ai montré comme étant « une protéine » du SARS-Cov2 n’est en fait qu’un domaine, c’est à dire un morceau de protéine. La compétition CASP porte d’ailleurs souvent seulement sur des domaines et pas des protéines entières, car on considère que les différents domaines d’une protéine se replient suffisamment indépendamment les uns des autres pour pouvoir les traiter plus ou moins séparément.
D’ailleurs j’ai choisi de ne pas introduire ces termes, mais pour les protéines on parle de structure primaire (pour les chaines), secondaires (avec les hélices et feuillet), tertiaire (pour la forme complète) et quaternaire (pour les interactions avec les autres protéines). Vous verrez peut-être passer souvent ces termes.
Concernant les maladies provoquées par un mauvais repliement, j’ai mentionné le cas de certaines maladies neurodégénératives causées par des substances amyloïdes. Si j’ai bien suivi, dans ce cas là ça n’est pas une perte de fonction (liée à un mauvais repliement) qui pose problème, mais l’accumulation en dépôts fibreux (plaques) qui causent des perturbations dans les tissus.
Structure et géométrie des chaines
J’ai passé sous silence la manière dont les acides aminés se lient pour effectivement former une chaine. Il s’agit de la liaison peptique, qui permet d’accrocher l’acide d’un AA avec l’amine d’un autre.
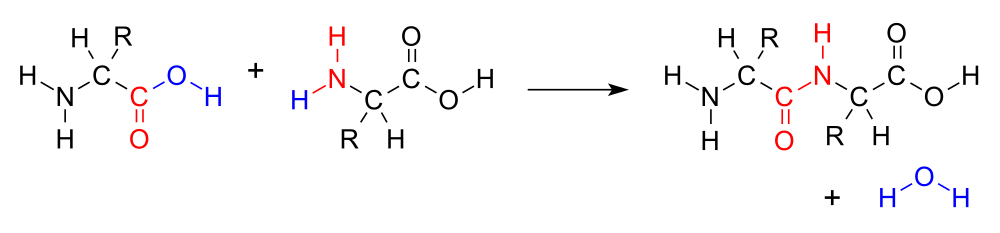
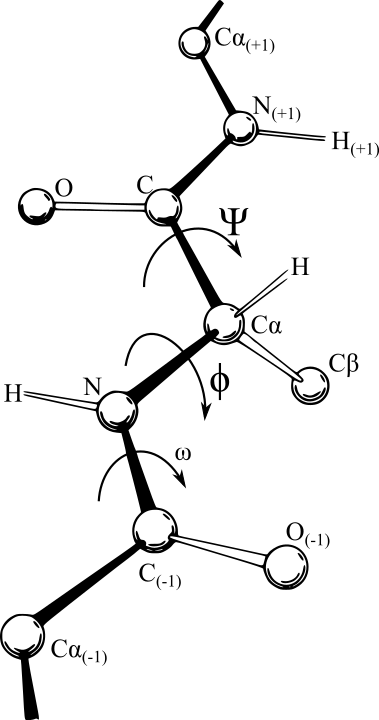
Un point intéressant concerne la géométrie spatiale qu’on obtient. Ci-dessous, vous avez une image du squelette de la chaine. On voit un acide aminé au milieu, relié par une liaison peptique à son prédécesseur et son successeur. Pour caractériser la géométrie du squelette, il faut donner les angles Phi et Psi de chaque acide aminé, ainsi que l’angle associé à la liaison peptidique (noté ici \(\omega\)). La nature de cette liaison fait que cet angle est contraint à 0 ou 180°, et donc les degrés de liberté restants ne concernent que les angles au sein de chaque AA (dits « dihédraux »)
 Il existe des représentations permettant de visualiser les distributions des valeurs angles dihédraux. (Encore merci Wikipédia). On appelle ça un diagramme de Ramachandran
Il existe des représentations permettant de visualiser les distributions des valeurs angles dihédraux. (Encore merci Wikipédia). On appelle ça un diagramme de Ramachandran
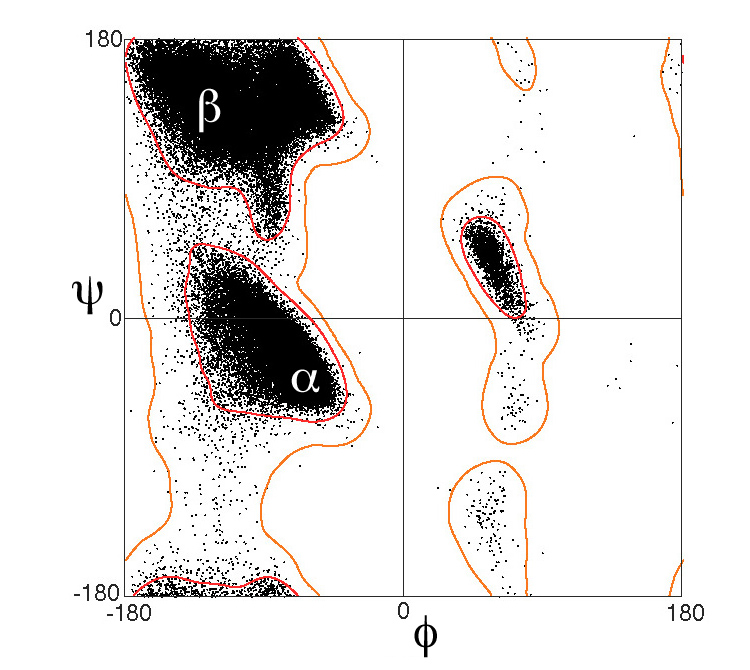
Sur celui ci-dessus, on voit clairement des concentrations dans certaines régions qui correspondent aux hélices alpha et aux feuillets beta.
D’ailleurs concernant les hélices, j’ai mentionné le cas le plus courant d’une liaison avec l’AA situé 4 cases plus loin, mais les cas +3 et +5 existent aussi. On trouve d’ailleurs un mélange puisqu’en moyenne une hélice alpha est en fait une hélice 3.6 (la valeur moyenne du pas.)
La cristallographie aux rayons X
Voici un sujet qui aurait mérité plus d’approfondissement, car il est à la fois très technique et très important. Notons que déjà un certain nombre de prix Nobel ont déjà été distribué pour la résolution de structures de protéines par cette technique.
L’étape de cristallisation semble particulièrement ardue : il faut purifier, mettre dans un solvant, trouver des conditions de cristallisation. Un biochimiste avec qui j’en ai discuté (merci Antoine !) me disait qu’il testait typiquement entre 600 et 1000 conditions de cristallisation différente pour espérer en trouver une qui fonctionne !
Ensuite sur l’interprétation de la figure de diffraction, il s’agit en fait d’un processus itératif faisant intervenir des modélisations que l’on va ensuite fitter aux données expérimentales.
Concernant d’ailleurs les simulations que j’ai évoquées, et l’histoire des niveaux d’énergie, le paramètre que l’on cherche à minimiser est l’énergie libre, qui prend à la fois en compte l’énergie et l’entropie. En pratique, comme on est pas au zéro absolu, ce qu’on va avoir est plutôt une distribution de niveaux d’énergie.
La compétition CASP
Je n’ai pas tout regardé des résultats de la compétition CASP, mais tout est en ligne. On trouve notamment des diapos de la conférence qui vient d’avoir lieu et au cours de laquelle les résultats ont été annoncés.
On y trouve d’ailleurs des réponses à une question qu’on peut légitimement se poser : est-ce que par chance la compétition de cette année était « plus facile » que les autres années, ce qui pourrait expliquer le score faramineux de DeepMind ? Eh bien a priori non.
On peut caractériser la difficulté en regardant à quelle point les domaines proposés étaient éloignés de chose connues, et voici le résultat en comparant aux années précédentes (pour les 3 catégories)
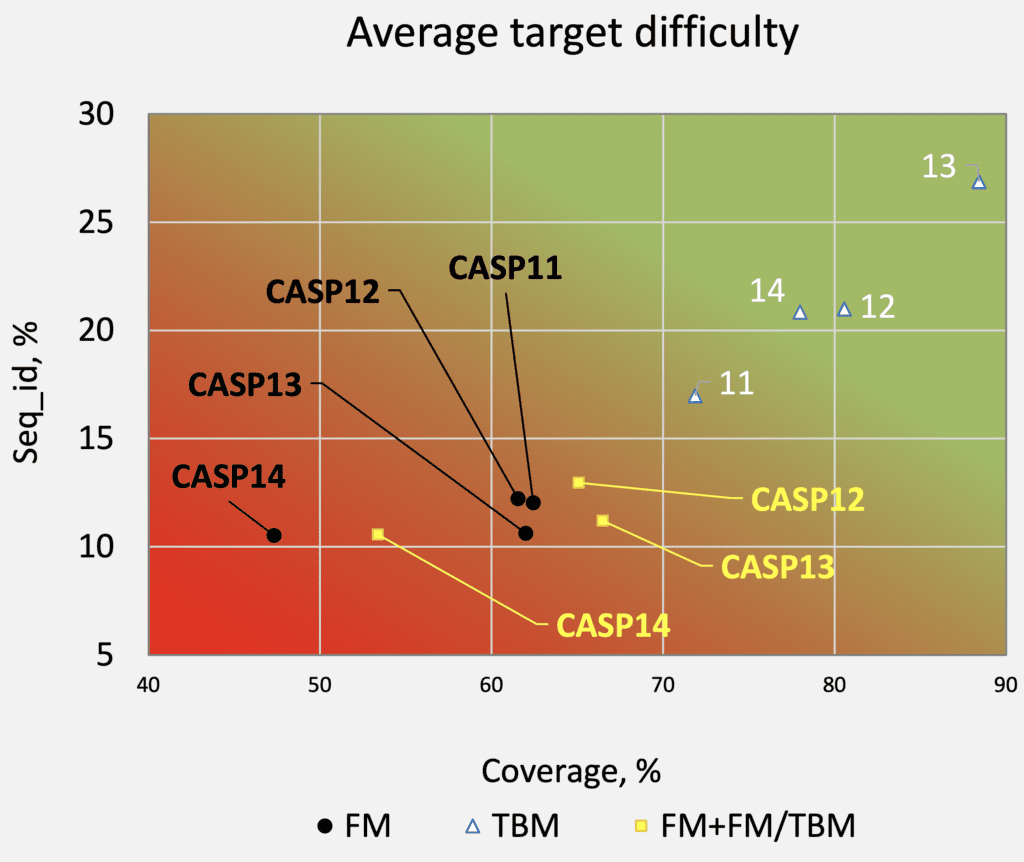
On voit que la compétition CASP14 de 2020 est a priori la plus difficile de ces dernières années.
Malgré tout, et c’est important de le noter, la performance en prédiction d’AlphaFold2 est évidemment dépendante du contenu des données utilisées pour entrainer les neurones. Si on s’amusait à inventer ex-nihilo des protéines (de façon totalement synthétique) très différentes de celles existantes, il est vraisemblable que la performance ne serait pas si bonne. On ne peut pas dire qu’AlphaFold2 soit « universel » dans sa capacité de prédiction.
Le fonctionnement d’Alphafold
Pour parler des entrailles de l’algorithme, je me suis essentiellement basé sur le papier qui est sorti au début de l’année sur AlphaFold 1
Senior, Andrew W., et al. « Improved protein structure prediction using potentials from deep learning. » Nature 577.7792 (2020): 706-710.
Ainsi que sur le billet de blog sorti par DeepMind pour CASP14. Entre temps, j’ai aussi vu les diapos de cette présentation à la conférence CASP.
Dans le papier, on a une architecture présentée qui est celle dont j’ai surtout parlé
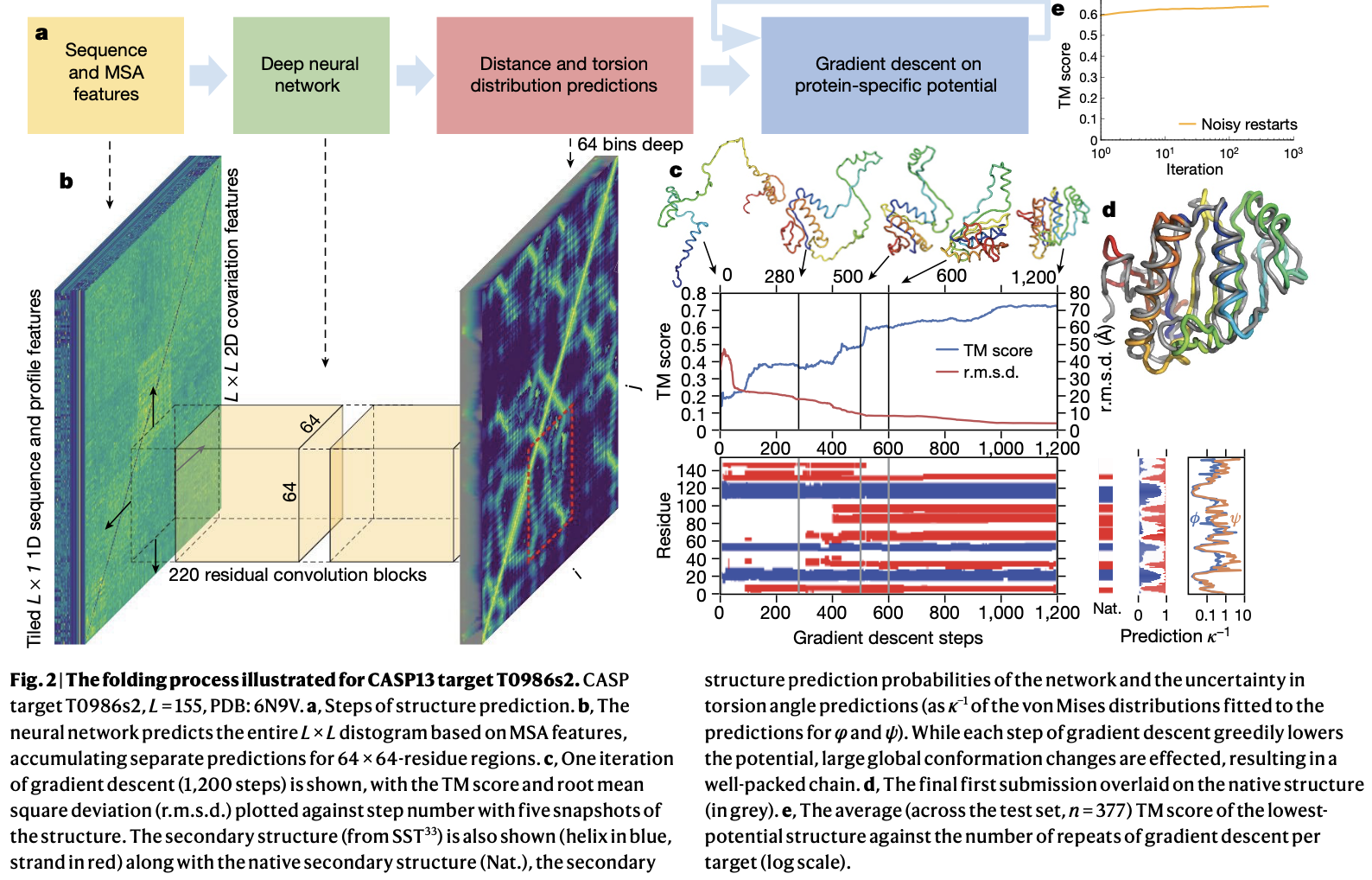
Comme je le dis dans la vidéo, l' »image » en entrée est très riche, puisque pour chaque paire d’acide aminés, on a plus de 480 features. Deux points à noter : en plus de la matrice de distance, on prédit également les angles dihédraux, et surtout ce ne sont pas des prédictions « uniques » mais des distributions.
On peut le voir dans le papier, qui l’illustre sur une matrice. On regarde l’acide aminé n°29 (représenté par une seule ligne/colonne dans la matrice), mais derrière ça il y en fait une distribution de probabilité pour la distance à chacun des autres AA.
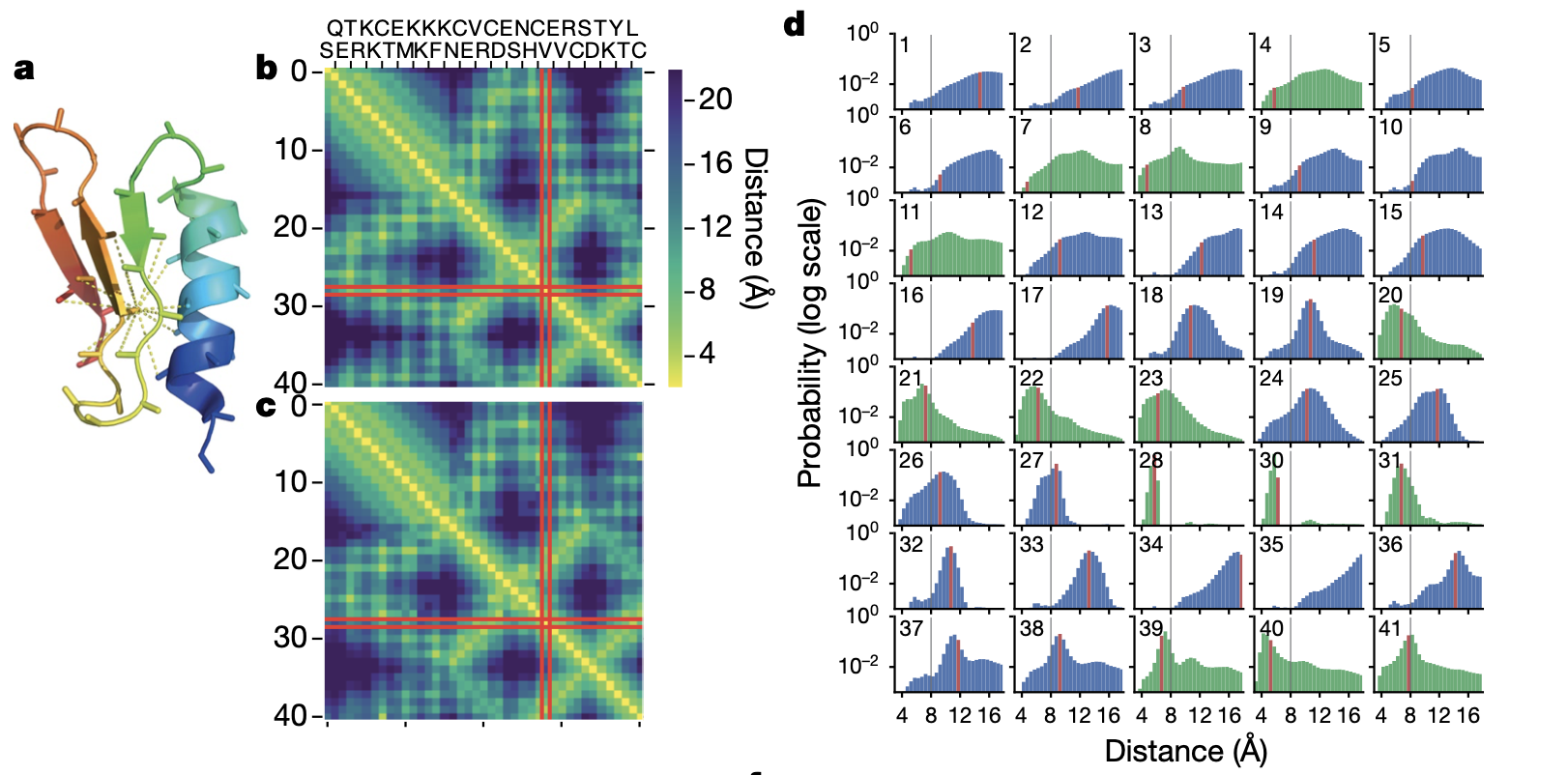
Le travail de reconstruction de la forme à partir de la matrice de distance est en fait une maximisation de probabilité.
Tiens notez au passage que sur la matrice de distance, on « voit » bien les structures secondaires : l’hélice alpha ce sont les AA de 1 à environ 14, où chaque AA est peu éloigné de ses voisins. Les feuilles beta ce sont les petites « anti diagonales »
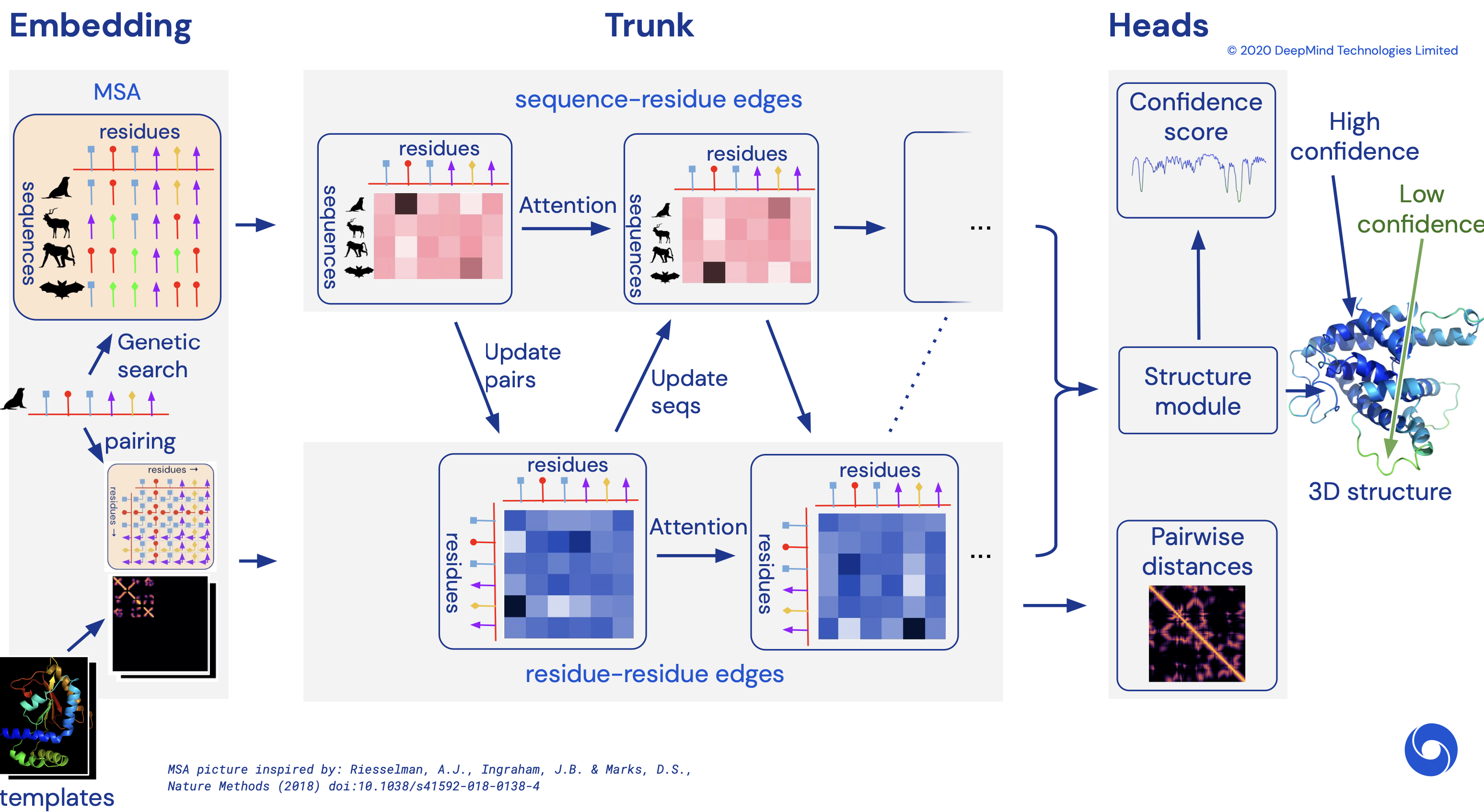
Bon mais tout ça c’était AlphaFold 1, qu’est-ce qui a changé avec AlphaFold 2 ? On ne le sait pas complètement. L’architecture que semble adopter AlphaFold 2 semble potentiellement un peu différente (ci-dessous un extrait de la conférence CASP)
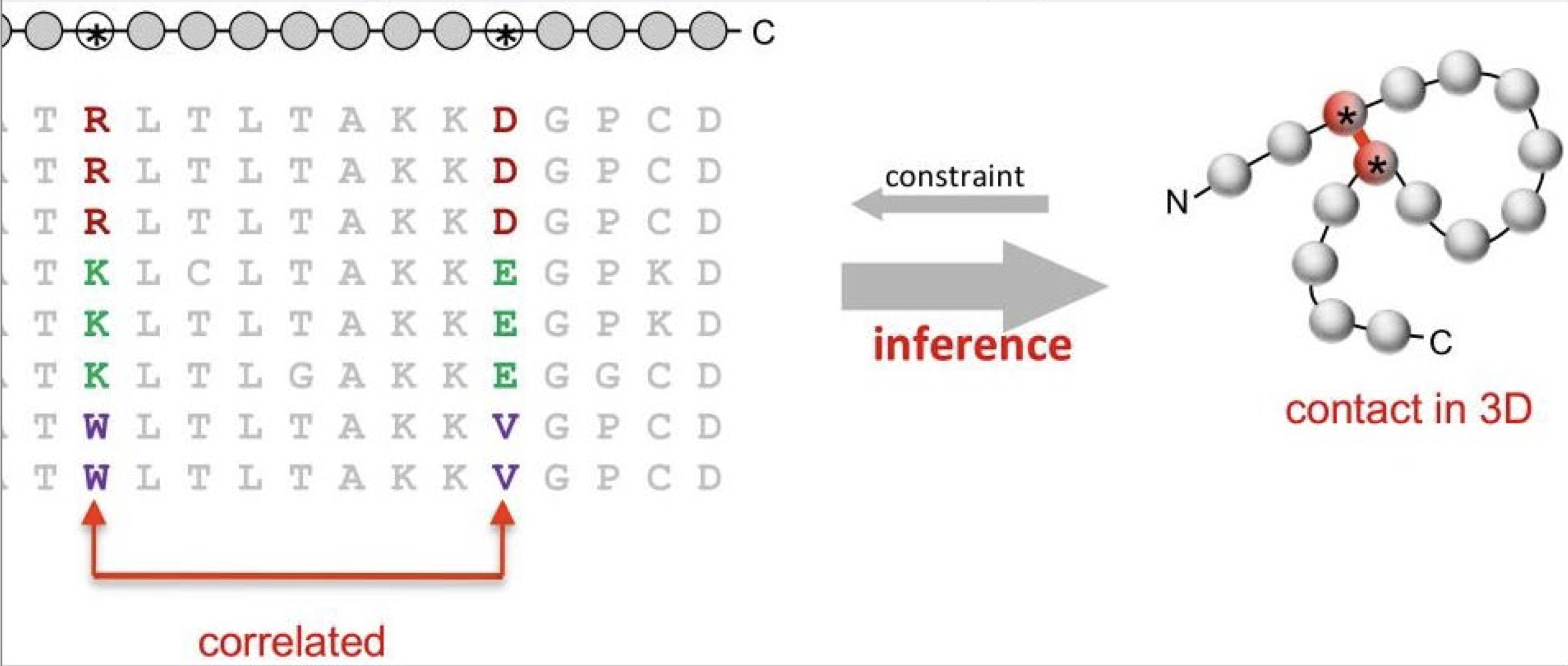
Une idée intéressante qui mérite d’être développée, c’est celle des « MSA » : Multiple Sequence Alignement (bien qu’elle ne soit pas spécifique au travail de DeepMind). L’idée consiste pour une séquence donnée à rechercher dans les bases de données des séquences qui ressemblent (par exemple codant pour un gène proche chez une autre espèce), et à voir quelles sont les AA qui soit ont été préservés, soit ont co-évolué. Quand deux AA varient toujours ensemble, cela laisse penser que changer l’un sans changer l’autre n’est pas viable, et donc que ces deux AA doivent avoir un rôle structurel lié. C’est ce qu’on trouve dans l’image ci-dessous (extrait toujours des diapos de la conférence)
Toujours sur AlphaFold, une question qu’on peut se poser concerne le temps (et le nombre de processeurs) nécessaires pour le faire tourner. Apparemment, pour la partie « entrainement », on parle de 128 TPU (Tensor Processing Unit) pendant quelques semaines. C’est beaucoup, mais évidemment pas quand on a Google derrière. Pour la partie prédiction, c’est moins clair. Sur du deep learning classique (un réseau utilisé en feed forward), la prédiction va très vite. Là ça semble plus compliqué. Il semblerait que DeepMind ait été évasif là-dessus pour l’instant. Ca serait plutôt de l’ordre de plusieurs heures, en tout cas pas « quelques millisecondes ».
Donc on peut se demander si, pour la conception de nouveaux médicaments, AlphaFold peut être pour l’instant compétitif face à des approches expérimentales « haut-débit » qui consistent à tester des tas de molécules de façon parallèle. Je ne connais pas assez la question pour répondre !
Merci à Antoine Schramm du Max Planck Institute, qui a répondu à mes questions naïves et tordues !




47 Comments
Passionnant ! Tu évoques au début de la vidéo que cette découverte pourrait aider à la lutte contre le changement climatique, quel est le lien avec les protéines ?
Merci !
J’avais lu des choses sur des protéines pour refixer du carbone, il faut que je retrouve la réf 🙂
Potentiellement, on pourrait modifier l’efficacité de la RuBisCO (ou plus globablement de la machinerie photosynthétique) 🙂
Convergence for a single protein estimated to take « a matter of days », according to the DeepMind blog post.
Estimated to be about $10,000 of compute time, if you were to rent processing capability from Google of the sort they were using.
Compare the recent Image-GPT algorithm for image completion, which also was based on transformer attention units, and also took a lot of processing to execute each task.
Impressionnant, très d’actualité en plus avec le COVID ça m’fait m’poser plusieurs questions sur est-ce que c’est du fait qu’on ne connait que très peu le repliement protéique qu’on a fait un vaccin à base d’ARN n’étant pas capable de créer un médoc protéique ?
Bonne continuation à toi
Coucou, je me permets de répondre (doctorant en virologie) ! La réponse rapide est : Non.
Et de manière plus détaillée : les vaccins à base de protéine sont nombreux, tout comme les médicaments (je fais personnellement la distinction ; les vaccins étant prophylactiques, donc agissant avant une maladie, et les médicaments étant justement pour agir sur une maladie déjà là). Par exemple le vaccin contre l’hépatite B est protéique, et l’insuline est un des exemples de protéine médicament.
L’astuce étant, comme dit dans la vidéo, que les cellules produisent les protéines à partir de l’ADN, et que ces dernières se replient d’elle même (ou parfois à l’aide d’autre protéines de la cellule). Donc pour les vaccins/médicaments protéiques, il suffit juste de faire produire la protéine in vitro à des cellules (des bactéries, pour l’insuline par exemple), puis de la purifier. Ainsi, pas besoin de connaitre la structure, juste de les produire et de les récupérer.
L’usage du vaccin ARN dans le cas du Covid, est à mon sens (mais ce n’est qu’une hypothèse raisonnable à partir des mes connaissances, je ne suis pas aller vérifier) justement car ils sont beaucoup plus facile à produire que les vaccins protéiques. Certaines protéines une fois produite in vitro par les cellules d’intérêt sont très difficile à purifier, tandis que l’usage chez l’homme de protéines produites in vitro est (heureusement) soumis à des impératifs de pureté très exigeants. Ces mêmes protéines peuvent aussi pour diverses raisons être difficiles à produire, avant même de passer à leur purification. Ainsi avec les vaccins ARN on évite toute cette lourdeur, le plan de construction de la protéine d’intérêt est fournit à nos cellules, elles se débrouillent pour les produire elle même, puis les protéines peuvent servir à « l’éducation » du système immunitaire.
Je ne suis pas encore assez au fait de la technologie détailler de ces vaccins ARN, mais après coup je suis plutôt surpris qu’ils n’arrivent que maintenant, les besoins de leur mise au point étant à priori assez légère (fondamentalement la séquence ADN d’une protéine x ou y doit suffire à obtenir une séquence ARN utilisable en vaccin).
Malgré tout, la connaissance des structures 3D des protéines reste quelque chose d’important, et permet/permettra notamment de modéliser/cibler les interactions entre une protéine donnée et un médicament.
Excellente vidéo !
Je me demande si l’on est sûr qu’une protéine est assurée d’avoir une unique forme. Par exemple serait il possible que lors de son repliement elle tombe dans un minimum local d’énergie ?
Bonjour,
Je me permet de répondre à la question, ayant fait une thèse sur un sujet connexe.
La réponse est non, les protéines n’ont pas une forme unique. Tout d’abord, si on tire une séquence d’acide aminés au hasard, il y a de très fortes chances que la protéine associée ne se replie pas du tout. Les protéines qu’on étudie dans CASP sont présentes dans des organismes vivant, et ont donc été sélectionnées par l’évolution pour se replier de manière plus ou moins reproductible pour assurer leur fonction. Cependant, on peut apporter des nuances, d’une part la protéine peut cristalliser dans une conformation différente de celle qu’elle adopte en solution, d’autre part, il y a souvent des parties rigides et des parties plus flexibles dans la protéine, les parties flexible ayant plusieurs conformations.
Salut! Très intéressant 🙂 Cependant, est-il possible d’avoir accès à tes références ?
Pardon, je viens de comprendre qu’elles sont au fur et à mesure du texte. Merci!
Oui je n’ai pas présenté ça de façon très organisée j’avoue, il faudrait que je regroupe à la fin 🙂
Très bon bonne vidéo, très bon billet. Juste quelques remarques :
A 2″37, une version plus réaliste de l’acide aminé serait de l’écrire avec ses extrémités NH3+ et COO-, bon c’est juste un détail 😉
A 5″12, ce n’est pas exactement une représentation avec « tous » les atomes, il manque les H, puisque 99% du temps ils ne sont pas visibles en cristallographie. On peut d’ailleurs les générer très facilement avec certains programmes, et ça change quand même l’aspect et le volume de la molécule qu’on regarde.
Enfin, pour les maladies neurodégénératives et les amyloïdes, on pense que c’est bien l’accumulation en plaques qui cause des problèmes, mais il semble qu’il y a AUSSI un « mauvais » repliement de la protéine associé (et qui serait lié à cette accumulation justement). C’est assez bien documenté sur les maladies à prion par exemple, avec les 2 formes de la protéine PrP, PrPC et PrPSc. Quoique le debat ne soit pas (à ma connaissance) entièrement tranché.
Bref, sinon c’était très bien, merci pour cette video !
Pour les amyloïdes
« Ca serait plutôt de l’ordre de plusieurs heures »
Dans la vidéo tu racontes qu’en 1/2 heure le gars avait la réponse. Donc on est plutôt sur de la dizaine de minutes, non ?
Apparemment lors de la conférence CASP, lors des questions orales ils ont été évasifs et répondu « quelques heures ».
Je ne sais pas si l’histoire de Lupas est représentative, peut-être qu’ils ont mis les bouchées double cette fois là car c’était un jury de CASP ? Peut-être qu’il déforme / exagère dans l’interview ?
Non, pour l’histoire de la demi-heure, ce n’est pas le temps qu’il a fallu à Alpha Fold pour trouver la structure,, c’est le temps qu’il a fallu au cristallographe pour vérifier si la réponse d’Alpha Fold était bien compatible avec ses clichés de diffraction. C’était une structure inconnue que même eux n’arrivaient pas à résoudre malgré leurs expériences, ils ne connaissaient donc pas le résultat.
Disons pour faire simple que l’étape « clichés de diffraction => structure » peut etre compliquée car une part de l’information nécessaire est manquante, et il faut un peu ruser pour réussir cela, souvent en s’aidant à nouveau de structures déjà existantes. En revanche, si on connait la structure (ou une structure assez proche), il est nettement plus simple de vérifier que les clichés de diffraction sont bien en accord avec cette structure. D’apres ce que je comprends, c’est précisément ce qu’on fait Lupas et ses collègues pendant ces trente minutes.
Bonjour, il me semble que la demi heure evoquee concerne la verification par le jury de la prediction faite par l’IA. mais bon, je ne suis pas fort en anglais, j’ai peut etre mal lu, en faisant « pause » sur la video..
cdlt
Merci David pour toute cette clarté.
Ce qui me frappe, dans le repliement des protéines, c’est la transition entre l’information-flux et l’information-structure. Cette distinction avait été faite par Henri Atlan, a une époque où le mot information autorisait beaucoup de délires non-scientifiques. Si une protéine est une séquence linéaire, comparable à l’axe syntagmatique d’un texte, le repliement de la protéine sur elle-même en fait une structure, un objet, voire une machine en trois dimensions, capable de lire un texte. Mais aussi capable de laisser passer ou non un autre objet, en fonction de sa structure. D’où la métaphore que tu emploies, de la clef et de la serrure. C’est pourquoi la compréhension des règles de ce repliement serait une révolution.
Super intéressant ! Je suis tout d’abord tombé sur ta vidéo et pleins de questions me sont venus en tête !!
Est-il possible que le GDT soit référencé par l’IA et que les méthodes scientifiques avec laser soit misent en compétition ?
Aussi, à un moment tu parles très rapidement d’un autre sujet que je trouve super intéressant, celui des protéines mal formées qu’on retrouve chez les personnes atteintes d’alzheimer. Est-il possible de les réformer en s’appuyant sur les résultats obtenus grâce aux IA ? je sais pas si c’est très claire ^^` et je vais m’arrêter là pour les questions, mais en tout cas, si j’ai une réponse à une de ses 2 questions je serait déjà super content !! =D
Il est 1 heure du matin, j’ai fumé un gros pétard et boudu… j’apprends qu’on est pas loin d’avoir réussi à cracker la clé de la vie. Je suis impatient de savoir ce que j’apprendrais dans 20 ans
Une fois qu’on connaît la forme de la chaîne d’acide-aminé que peut on en déduire ? Sa forme permet-elle de connaître sa fonction ?
Je me posais la même question, on améliore notre connaissance de la forme, mais comment peut on savoir ce a quoi sert la forme. Est on condamné au constat expérimental ou commence t on a faire des prédictions ?
Bonjour,
je tiens à vous féliciter pour votre travail et votre vulgarisation extraordinaire ! Vos vidéos sont vraiment passionnantes, et puis, bientôt le million d’abonnés. Vous le méritez bien avec tout ce travail.
Merci !
Je me permets de me joindre aux félicitations. Je suis toujours bluffé par la qualité de votre travail, c’est toujours clair et compréhensible, et je constate lorsque les vidéos traitent des sujets que je connais que la vulgarisation est toujours bien faite, avec un sujet rendu accessible sans pour autant tomber dans le faux. Et c’est une travail d’autant plus bluffant que vous passez d’un champ/sujet à un autre sans sourciller… Bien que je ne les connaisse pas absolument tous, et qu’il y a de la sérieuse concurrence (Coucou à Léo Grasset, Viviane Lalande et consort..) je dirais sans hésiter que vous être le (ou en tout cas l’un des) meilleur vulgarisateur français sur youtube.
Ou tout simplement en un mot : merci.
Trés intéressant.
Le développement de nouveaux médicaments s’est fait trés rarement à partir d’un criblage sur une protéine purifiée mais pratiquement toujours à partir d’un criblage fonctionnel (cellule our organisme comme le poisson zebre ou c.elegans).
La raison est probablement que dans la cellule on a des architectures moléculaires. Il faut interagir avec ces architectures moléculaires et pas seulement avec des protéines purifiées.
Par ailleurs, la concentration en eau dans un tube à essai est de 55M alors que dans la cellule en moyenne, on est autour de 30M, ce qui change énormément le comportement des protéines.
Excellente vidéo qui explique très très bien cette problématique captivante. Je me permets juste une petite remarque, en général on considère qu’il y a 20 acises aminés et pas 21, et l’analyse de la séquence de protéines dans les bases de données permet de constater qu’il y a en général 20 lettres seulement associées aux 20 acides aminés naturels. Celui qui semble être en trop dans la vidéo est la selenocysteine, U dans la vidéo.
C’est un acide aminé modifié, qui n’est pas pris en compte dans les prédictions de structure actuellement. Et il en existe d’autres, en effet beaucoup d’acides aminés peuvent être modifiés notamment par ce que l’on appelle des modifications post-traductionnelles, comme la phosphorylation, différents types de glycosylation, des acetylations, methylation, etc…
Ces modifications peuvent avoir une incidence très forte sur le repliement des protéines et rajoutent une couche de complexité supplémentaire, et qui sait, peut être qu’un alphafold 3 ou 4 ou plus sera capable d’affiner les structures des protéines en fonction de la présence de ces modifications. En tout cas c’est déjà une sacrée prouesse et un bond en avant notable dans le domaine
Merci pour la précision sur la sélenocystéine ! J’ignorai qu’on ne la prenait pas en compte dans la prédiction.
Un grand bravo et merci pour cette vidéo passionnante et si bien expliquée. En tant que biologiste j’ai tilté sur le nombre 21 d’acides aminés constituant les protéines de tous les êtres vivants. Je pensais que ce n’était que 20 et finalement, sur Wikipedia, on trouve qu’il y en aurait 22 qui soient « protéinogène » car la pyrrolysine et la sélénocystéine seraient un peu des cas à part (https://fr.wikipedia.org/wiki/Acide_amin%C3%A9_prot%C3%A9inog%C3%A8ne). Mais bon c’est vraiment un détail qui n’enlève en rien au talent et à la prouesse mis en œuvre pour nous expliquer si bien cette histoire de plus 50 ans qui semble avoir trouvé un dénouement impressionnant d’efficacité et de rapidité pour la détermination du repliement des protéines !
Bonjour,
Cette vidéo est une excellente vulgarisation des protéines.
Mon plaisir a été un peu terni par votre utilisation de l’Angstrœm qui a été exclu par la convention internationale des poids et mesure en 1960 lors de sa définition du système international d’unités (SI).
Bravo encore pour votre travail.
Merci ! Ca n’est pas une unité que j’utilise dans la vraie vie, mais pour de la vulgarisation, c’est pratique d’avoir une unité qui soit « de l’ordre de la taille d’un atome » !
@Charles: Dans le domaine, l’angstroem est utilisé comme unité principale. Désolé de pas respecter le SI, j’espère que vous vous en remettrez 😉
@David: Je suis impressionné par la qualité scientifique de la vidéo!
Bonjour,
Super vidéo comme d’hab !
Je viens de regarder ta vidéo, ainsi que tes deux vidéos sur la thermodynamique et je me posais une question. Tu dis, selon les principes de la thermodynamique, que ‘les systèmes essayent d’optimiser leurs énergies ». Or, la théorie du handicap me semble affirmer le contraire, et le processus évolutif parfois (ex : le nerf laryngé récurrent de la girafe). Ma question est donc la suivante : existe-t-il, à l’échelle moléculaire, des protéines, qui en raison du processus évolutif, ne tendent pas vers une optimisation d’énergie ?
Je suis pas très clair, mais je pourrais reformuler la question si besoin.
@Mat: « Existe-t-il, à l’échelle moléculaire, des protéines, qui en raison du processus évolutif, ne tendent pas vers une optimisation d’énergie ? » Oui, il en est question dans la vidéo, celles qui sont impliquées dans Alzheimer ou dans les maladies à prion type Creuzfeld-Jacob par exemple. Les protéines fonctionnelles sont dans un minimum local d’énergie (pas un minimum global donc), mais sous certaines conditions elles peuvent passer dans leur forme optimale d’énergie (minimum global), dans laquelle elles ne sont plus fonctionnelles.
En ce qui concerne l’hypothèse du handicap, ça n’a rien à voir, puisque cette hypothèse s’occupe de l’évolution des êtres vivants, et les protéines ne sont pas des êtres vivants…
GG pour le million! Tu le mérites amplement et j’espère que tu as toujours autant de plaisir à faire des vidéos!
Très bonne vidéo, très bien structurée et vulgarisée. Quelques commentaires tout de même, étant moi-même biologiste/structuraliste:
« Il ne suffit pas de mettre sa protéine sous le microscope pour la voir ». Et bien si! La technique phare en biologie structurale utilisée partout et qui a très récemment atteint la résolution atomique c’est la cryo-electron microscopie (prix Nobel de 2017), et on met littéralement sa protéine (ou son complexe de protéines) sous le microscope. C’est une méthode qui est entrain de/a dépassé la cristallographie en terme de rapidité et d’efficacité, notamment car il n’est pas nécessaire d’obtenir des cristaux de protéines et aussi et surtout car elle permet d’étudier des complexes protéines/protéines, protéines/acide nucléiques petits ou de très grande taille de manière facile et en conditions quasi natives! Elle fait quasi tout en mieux que la cristallographie, si tant est qu’on ait le bon microscope.
La prouesse d’AlphaFold2 reste très impressionnante, mais a prouvé son efficacité uniquement sur de petites protéines ou des domaines isolés (partie de protéine). Et comme expliqué, les protéines ne sont jamais seules dans la cellules, elles interagissent ensemble pour former des complexes entres-elles ou avec d’autres molécules et c’est là ou réside la vraie fonction des protéines, et ça, pour l’instant, aucun logiciel n’arrive à le prédire.
AlphaFold2 permettra néanmoins de fournir de bon modèles initiaux pour l’étude de ces complexes, si le code est un jour rendu publique … chose qui fait largement débat dans le domaine en ce moment.
Merci encore pour cette vidéo et ce billet! Ca fait plaisir de voir quelque chose qui me touche directement abordé ici!
Et félicitations pour le million d’abonnés!
Pingback: Le repliement des protéines & l’I.A AlphaFold de DeepMind – David Louapre – Phil-Dev Deeplearning
Excellente vidéo à nouveau, toujours aussi émouvante.
Travail de vulgarisation d’autant plus bluffant que, quoi…, il s’est juste passé une bonne semaine entre l’annonce de l’exploit d’alphafold, et la publication de ta vidéo !?
Y’a une I.A. qui bosse à ton service ou quoi ?
Félicitations !
Beau boulot, super vidéo !
Concernant les limites d’AlphaFold 2 : http://occamstypewriter.org/scurry/2020/12/02/no-deepmind-has-not-solved-protein-folding/ (l’auteur est journaliste scientifique pour The Guardian)
Un des points important d’AlphaFold c’est qu’il utilise la co-évolution des acides aminés comme « input » (pour plus d’info, voir les travaux du Prof Paolo De Los Rios de l’EPFL – collègue^^).
Pour gagner en temps de calcul, il y a eu un choix de limiter le nombre de paramètres dispo avec ces inputs. Quelles sont les conséquences ? Et bien en prenant AlphaFold1 on a parfois des erreurs lorsqu’on aligne les structures avec la version crystalisée.
Pour le moment, et si je devais résumer je dirais « dans l’ensemble ça va mais on est encore loin d’avoir trouver LA méthode pour résoudre le repliement des protéines » + on ignore le détail de ce qu’à fait Google, il faudra attendre le papier.
Autre point: DeepMind c’est des dizaines (centaines?) de millions de dollars, des équipes attitrées uniquement à ce projet. Jamais des universitaires n’auraient pu faire cela… même en collaborant (ultra risqué, personne ne financerait cela).
Mais ce qu’ils ont fait est assez fort, en si peu de temps.
My2Cts.
Anthony – postdoc sur les chaperones/repliement des protéines 😉
Quelle est la raison qui fait qu’on ne peut pas « juste » mettre la chaine d’acide aminé dans un moteur physique qui décrit les liaisons et forces atomiques et laisser la simulation physique tourner ? C’est juste une question de temps de calcul trop long ou il y a des mécanismes physiques bas niveau encore non connus précisément qui empêchent cette approche ?
Je pense que c’est un mélange de plusieurs choses : d’une part pour que la simulation soit correcte, il faudrait une modélisation parfaite des champs de force, et je pense qu’on n’a que des choses imparfaites;
Ensuite on sait que le repliement lui-même est « assisté » (par exemple par des chaperones), donc il ne suffit pas de mettre la chaine en position initiale et la laisser se replier;
Hello,
merci pour la vidéo et pour l’article, le sujet est très intéressant!
Il y a quelque chose que je n’ai pas compris par contre: Comment on sait que la protéine a été repliée correctement ? Dans le cas d’une protéine inconnue, qu’est-ce qui permet de différencier un repliage correcte d’un autre qui serait faux, vu qu’on ne connait pas la réponse ?
merci d’avance
Salut! Étant étudiante en biologie je me permet de répondre : c’est par des méthodes expérimentales, notamment la cristallographie aux rayons X, que le repliement exacte d’une protéine est déterminée. mais ces méthodes sont très longues et couteuses, c’est d’ailleurs pour ça que réussir à déterminer précisément ces structures 3D seulement par des méthodes théoriques représenterait une révolution!
Merci pour cette video. J’ai beaucoup apprécié. J’aimerais en apprendre plus sur le problème de trouver les positions relatives de points (du plan ou de l’espace) à partir de la matrice des distances. Y a-t-il en général plus d’une d’une solution ? J’imagine que oui. Y a-t-il alors des conditions pour obtenir une solution unique ? Avez-vous des références sur ce sujet ?
Merci encore.
Pingback: I.A., une révolution par le jeu ? – CCSTIB
Bonjour,
Super chaine, supers vidéos, le titre de boss de la vulga fr est pas volé!
Sinon, je suis en train d’écrire un article de recherche sur un sujet connexe. Est ce qu’il est possible d’avoir la source du graph de GDP par an des meilleurs algo pour la catégorie free modeling de CASP svp (celui qui apparait dans la vidéo).
Merci,
Bonne journée
La voici : https://deepmind.google/discover/blog/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology/